3+3 doesn’t show 6
I have a problem which I don’t understand how to solve. I have this 2-bit adder, but when I select A1AO to be (11)2 3 and B1BO to be (11)2 3 and run it on Logisim it doesn’t show me 6. Instead it shows me 4.
How can I fix it?
2 answers
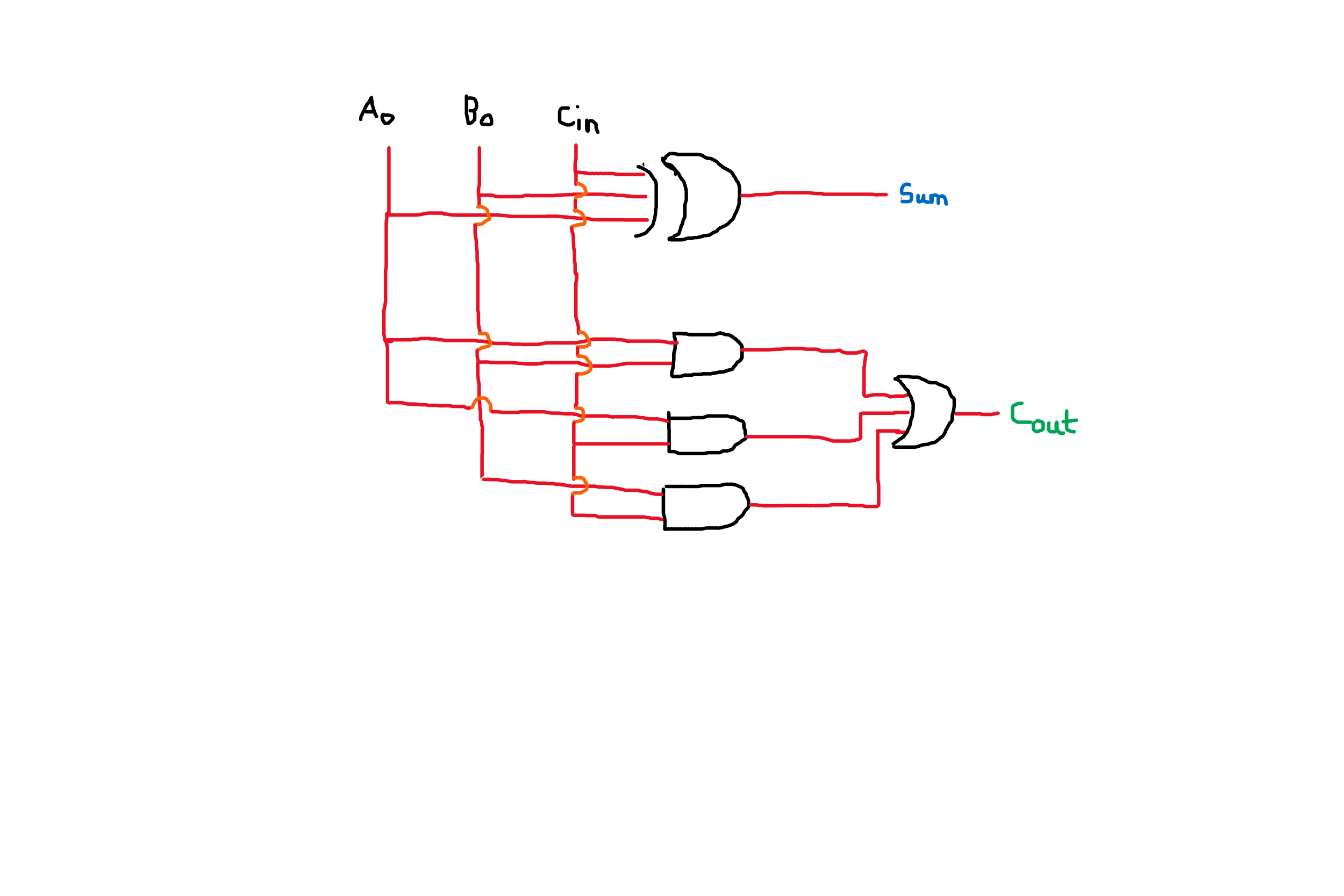
For the benefit of later readers, I thought I would explain why the original circuit "didn't work" and how exactly the "correct" circuit differs from the original circuit.
The issue is the 3-input XOR gate in the original circuit. The behavior of an XOR gate with more than 2 inputs is not precisely defined. Some people assume that a 3-input gate acts as F = A xor B xor C, which is the implementation in your "correct" circuit. However, some people define the XOR function as "1 and only 1", regardless of the number of inputs. If you used Logisim to simulate this circuit you discovered that Logisim defaults to the latter definition. As I recall, you can change the behavior of the gate in Logisim, but the default is "1 and only 1".
So, when all three inputs are 1 the Logisim default XOR will provide an output of 0. If you use two 2-input XOR gates the output will be 1, which is what you want in this case.


2 comment threads